Bio-painting
Logo design for Kavli Institute for Theoretical Physics (KITP) program



Logo design for Kavli Institute for Theoretical Physics (KITP) program

“Life Inspiring” Art Competition Winner (2023)
Art of Science Image Contest Winner (2024)
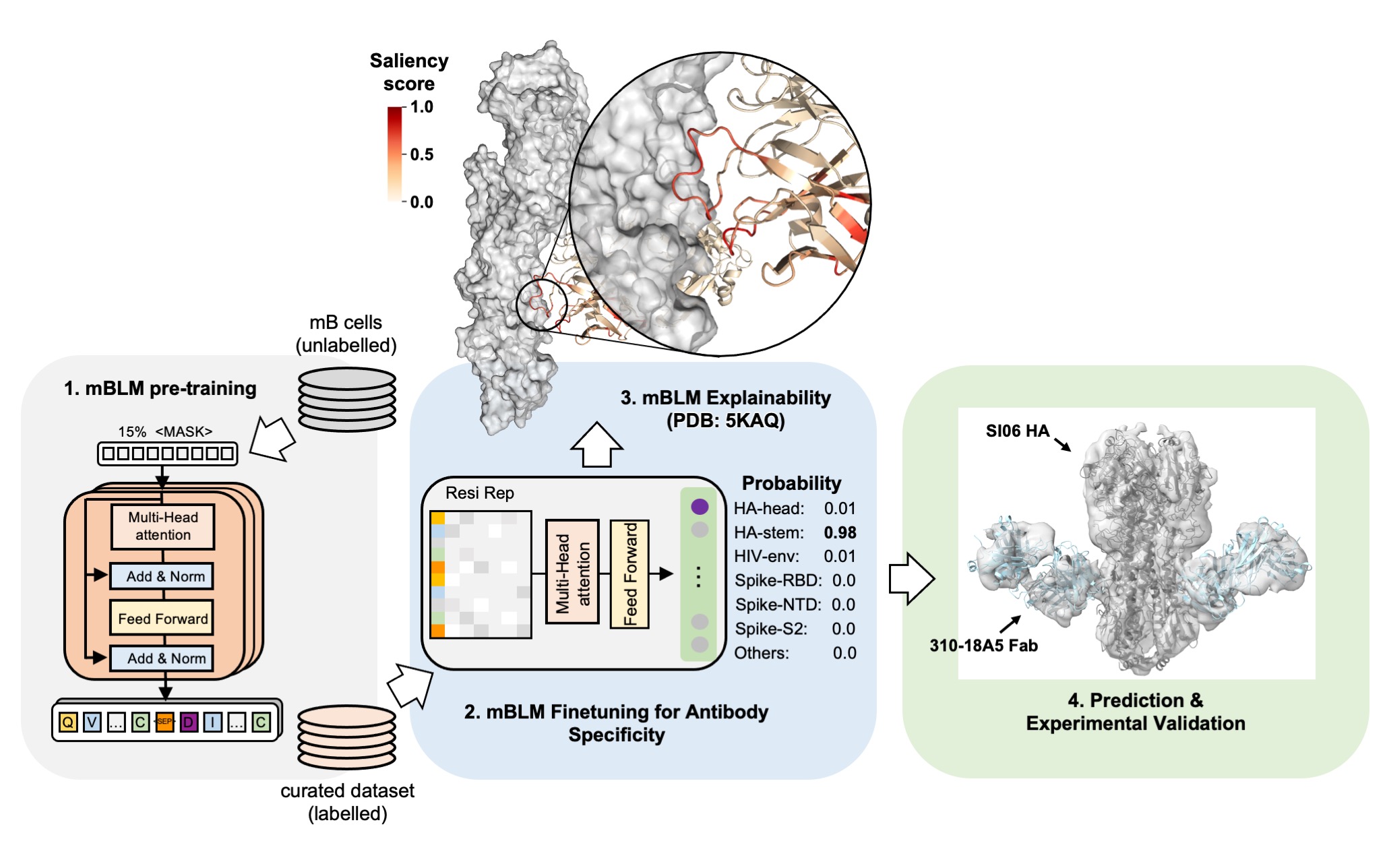
An explainable language model for antibody specificity prediction
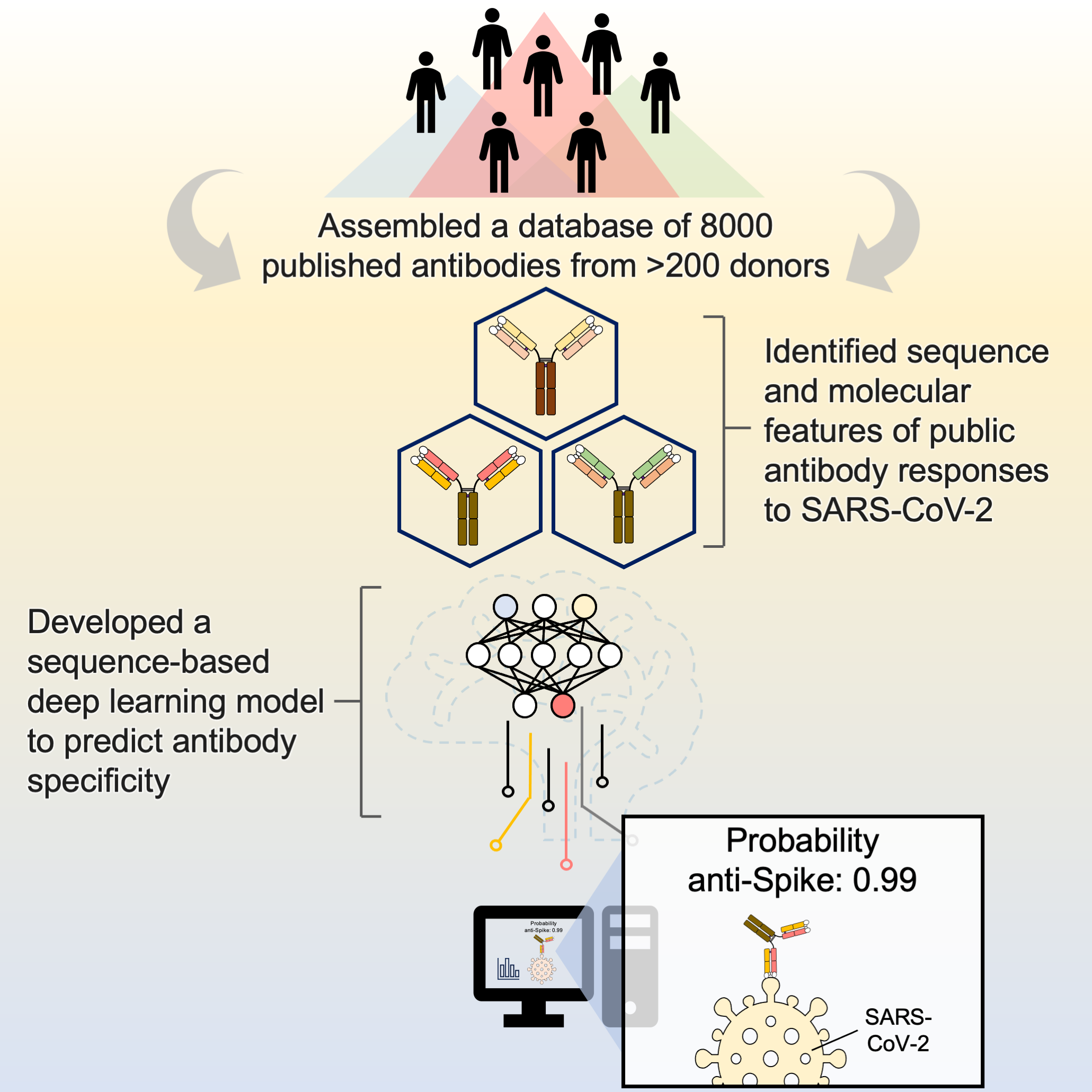
A large-scale systematic survey reveals recurring molecular features of public antibody responses to SARS-CoV-2
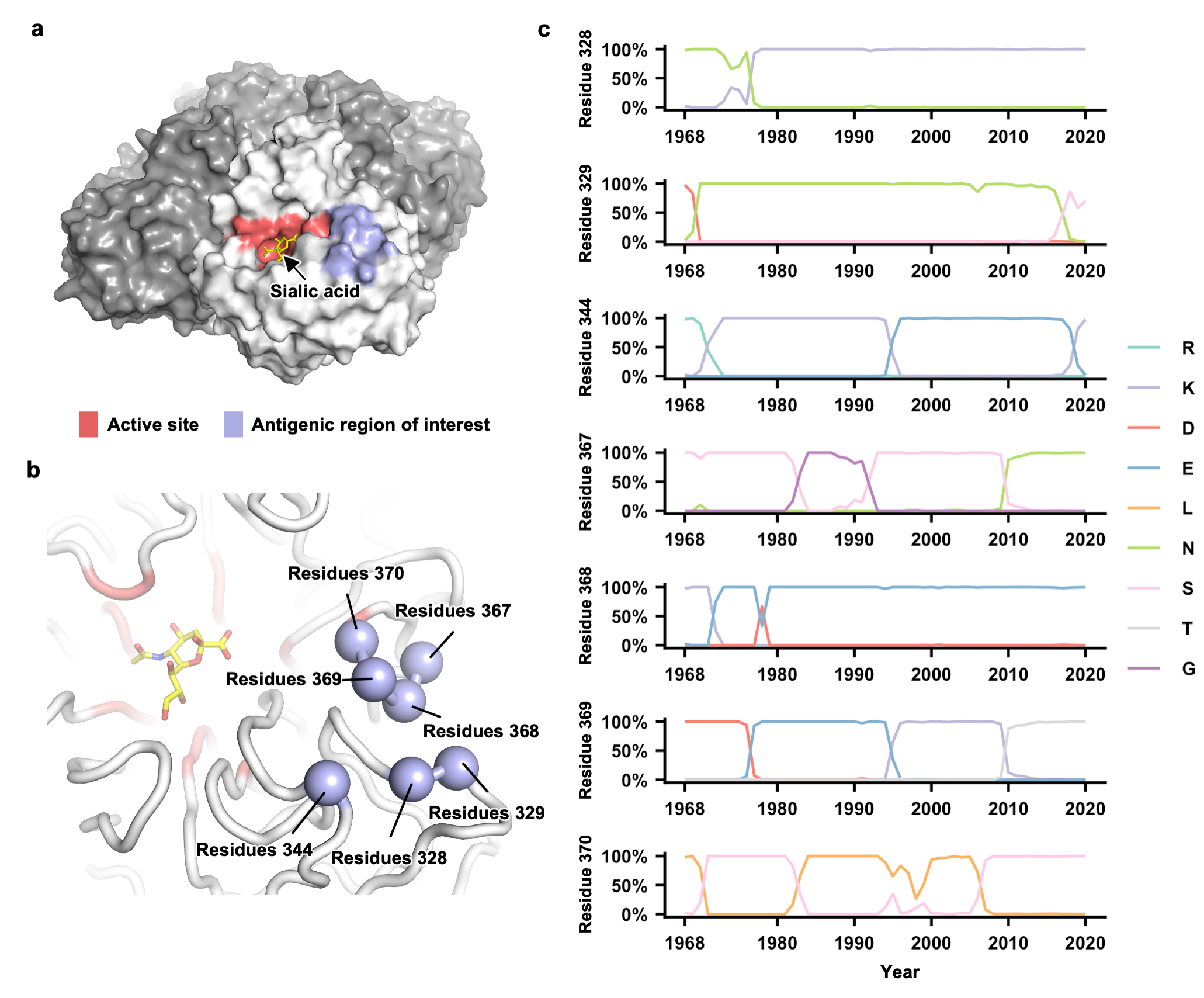
Antigenic evolution of human influenza H3N2 neuraminidase is constrained by charge balancing
[2025] Yiquan Wang has joined the University of Florida as an Assistant Professor. (Lab website )
[2024] Yiquan Wang Receives 2024 Outstanding Graduate Student Award for Exceptional Research and Innovation (link )
[2024] New AI model promises to speed up process of antibody characterization (link )
[2023] Winners announced for "Life Inspiring:" A gallery of images from the School of MCB (link )
[2022] Meet MCB: Biochemistry PhD student Yiquan Wang (link )
[2022] Machine-learning model can distinguish antibody targets (link )